

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
McEntyre J, Ostell J, editors. The NCBI Handbook [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2002-.

This publication is provided for historical reference only and the information may be out of date.

Summary
The protein database of Clusters of Orthologous Groups (COGs) is an attempt to phylogenetically classify the complete complement of proteins (both predicted and characterized) encoded by complete genomes. Each COG is a group of three or more proteins that are inferred to be orthologs, i.e., they are direct evolutionary counterparts. The current release of the COGs database consists of 4,873 COGs, which include 136,711 proteins (~71% of all encoded proteins) from 50 bacterial genomes, 13 archaeal genomes, and 3 genomes of unicellular eukaryotes, the yeasts Saccharomyces cerevisiae and Schizosaccharomyces pombe, and the microsporidian Encephalitozoon cuniculi. The COG database is updated periodically as new genomes become available. The COGs for complete eukaryotic genomes are in preparation. The COGs can be applied to the task of functional annotation of newly sequenced genomes by using the COGnitor program, which is available on the COGs homepage.
Introduction
The recent progress in genome sequencing has led to a rapid enrichment of protein databases with an unprecedented variety of deduced protein sequences, most of them without a documented functional role. Computational biology strives to extract the maximal possible information from these sequences by classifying them according to their homologous relationships, predicting their likely biochemical activities and/or cellular functions, three-dimensional structures, and evolutionary origin. This challenge is daunting, given that even in Escherichia coli, arguably the best-studied organism, only about 40% of the gene products have been characterized experimentally. However, computational analysis of complete microbial genomes has shown that prokaryotic proteins are, in general, highly conserved, with about 70% of them containing ancient conserved regions shared by homologs from distantly related species. This allows one to use functional information from experimentally characterized proteins to suggest function in their homologs from poorly studied organisms. For such functional predictions to be reliable, it is critical to infer orthologous relationships between genes from different species. Orthologs are evolutionary counterparts related by vertical descent (i.e., they have evolved from a common ancestor) as opposed to paralogs, which are genes related by duplication (1, 2). Typically, orthologous proteins have the same domain architecture and the same function, although there are significant exceptions and complications to this generalization, particularly among multicellular eukaryotes.
The COGs database has been designed as an attempt to classify proteins from completely sequenced genomes on the basis of the orthology concept (3–5). The COGs reflect one-to-many and many-to-many orthologous relationships as well as simple one-to-one relationships (hence, orthologous groups of proteins). In addition to the classification itself, the COGs Web site includes the COGnitor program, which assigns proteins from newly sequenced genomes to COGs that already exist and to several functionalities that allow the user to select and analyze various subsets of COGs.
Construction of the COGs
COGs have been identified on the basis of an all-against-all sequence comparison of the proteins encoded in complete genomes using the gapped BLAST program (6; see also Chapter 16) after masking low-complexity (7) and predicted coiled-coil (8) regions. The COG construction procedure is based on the simple notion that any group of at least three proteins from distant genomes that are more similar to each other than they are to any other proteins from the same genomes are most likely to form an orthologous set (Figure 1). This prediction holds even if the absolute level of sequence similarity between the proteins in question is relatively low, and thus the COG approach accommodates both slow-evolving and fast-evolving genes.
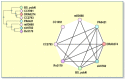
Briefly, COG construction includes the following steps:
- 1.
Perform the all-against-all protein sequence comparison.
- 2.
Detect and collapse obvious paralogs, i.e., proteins from the same genome that are more similar to each other than to any proteins from other species.
- 3.
Detect triangles of mutually consistent, genome-specific best hits (BeTs), taking into account the paralogous groups detected at step 2.
- 4.
Merge triangles with a common side to form COGs.
- 5.
Perform a case-by-case analysis of each COG. This analysis serves to eliminate false-positives and to identify groups that contain multidomain proteins by examining the pictorial representation of the BLAST search outputs. The sequences of detected multidomain proteins are split into single-domain segments, and steps 1–4 are repeated with the resulting shorter sequences, which assigns individual domains to COGs in accordance with their distinct evolutionary affinities.
- 6.
Examine large COGs that include multiple members from all or several of the genomes using phylogenetic trees, cluster analysis, and visual inspection of alignments. As a result, some of these groups are split into two or more smaller ones that are included in the final set of COGs.
By the design of this procedure, a minimal COG includes three genes from distinct phylogenetic lineages; protein sets from closely related species were merged before COG construction. The approach used for the construction of COGs does not supplant a comprehensive phylogenetic analysis. Nevertheless, it provides a fast and convenient shortcut to delineate a large number of families that most likely consist of orthologs.
The COGnitor Program
New proteins can be assigned to the COGs using the COGnitor program, the principal tool associated with the COGs database. COGnitor “BLASTs” the query sequences against all protein sequences encoded in the genomes that are classified in the current release of the COG system. To assign proteins to COGs, COGnitor applies the same principle that is embedded in the COG construction procedure, i.e., the consistency of genome-specific BeTs. For any given query protein, if the number of BeTs for a particular COG exceeds a predefined cut-off (three by default; the cut-off value can be changed by the user), the query protein is assigned to that COG; in cases where there are more than three BeTs to two different COGs, an ambiguous result is reported.
The Current State of the COGs Database, Updates, and Additional Classification of the COGs
Once the COGs have been identified using the above procedure, new members can be added using the COGnitor program. The assignments are further checked and curated by hand to eliminate potential false-positives. It has been shown that 95–97% of the COGnitor assignments typically require no correction (9). Once the proteins from a new genome are assigned to the appropriate pre-existing COGs through this combination of COGnitor and manual refinement, the remaining proteins from this genome are compared to the proteins from non-COG proteins from previously available genomes, and an attempt is made to construct new COGs using the original procedure. In addition, when new sequences are added to an exisiting COG, the COG is examined for the possibility of a split (isolation of a new COG) by inspecting BLAST search outputs for all COG members and, in some cases, phylogenetic tree analysis. Thus, the number of COGs continuously grows through the construction of new COGs that typically include just a small number of species, whereas the number of proteins in the COG system increases primarily through the addition of new members to pre-existing COGs.
In bacterial and archaeal genomes, approximately 70% of the proteins typically belong to the COGs. Because each COG includes proteins from at least three distantly related species, this reveals the generally high level of evolutionary conservation of protein sequences, making the COGs a powerful tool for functional annotation of uncharacterized proteins. The COGs were classified into 18 functional categories that loosely follow those introduced by Riley (10) and also include a class for which only a general functional prediction (e.g., that of biochemical activity) was feasible, as well as a class of uncharacterized COGs. A significant majority of the COGs could be assigned to one of the well-defined functional categories, but the single largest class includes the functionally uncharacterized COGs. Additionally, the COGs were clustered according to the common metabolic pathways and macromolecular complexes.
Phyletic Pattern Analysis in COGs
A phyletic pattern is the pattern of species that are represented or not represented in a given COG; alternatively, phyletic patterns can be described in terms of the sets of COGs that are represented in a given range of species. The COGs show a broad diversity of phyletic patterns; only a small fraction are universal COGs, i.e., they are represented in all sequenced genomes, whereas COGs present in only three or four species are most abundant. This patchy distribution of phyletic patterns probably reflects the major role of horizontal gene transfer and lineage-specific gene loss in the evolution of prokaryotes, as well as the rapid evolution of certain genes in specific lineages, which may be linked to functional changes. Phyletic patterns are informative not only as indicators of probable evolutionary scenarios but also functionally; most often, different steps of the same pathway are associated with proteins that have the same phyletic pattern, whereas on some occasions, complementary patterns indicate that distinct (sometimes unrelated) proteins are responsible for the same function in different sets of species. The COG system includes a simple phyletic pattern search tool that allows the selection of COGs according to any given pattern of species. This tool effectively provides the functionality of “differential genome display” (for example, allowing the selection of all COGs that are present in one, but not the other, of a pair of genomes of interest) and can be helpful for delineating sets of candidate proteins for a particular range of functional features, e.g., virulence or hyperthermophily.
Description of the COGs Website
The main COGs Web page contains the following principal features: (a) a list of all COGs organized by the (predicted) functional category; (b) separate lists of COGs for each functional category and for a variety of major pathways and functional systems; (c) a table of co-occurrences of genomes in COGs; (d) a list of COGs organized by phyletic patterns; (e) the phyletic patterns search tool; (f) the COGnitor program; (g) a search engine to search COGs for gene names, COG numbers, and arbitrary text; and (h) Help, which covers the principal subjects related to COGs.
The individual COG pages can be reached from any of the COG lists mentioned above or by searching the site (see, for example, the COG for exonuclease I). Each of the COG pages shows the respective phyletic pattern in a table that also gives the ID number for the contributing sequence(s), a cluster dendrogram generated using the BLAST scores as the measure of similarity between proteins, and a graphical representation of BeTs for the given COG (not shown for the largest COGs). Also, each of the COG pages is hyperlinked to: (a) pictorial representations of BLAST search outputs for each member of the COG, which also includes links to the respective GenBank and Entrez-Genomes entries (see, for example, the link from XF2022, the protein from Xyella fastidiosa in the exonuclease I COG); (b) a multiple alignment of the COG members produced automatically using the ClustalW program (11); (c) a FASTA library of the protein sequences that belong to the COG (represented by the floppy disc icon); (d) the respective functional category of COGs and pathway (functional system) if applicable (in this exonuclease I example, the functional category L represents proteins involved in DNA replication, recombination, and repair); (e) a COG information page that includes functional, evolutionary, and structural information on the COG and its members (many of these pages are still under construction); (f) other COGs that include distinct domains of multidomain proteins that belong to the given COG through one of their domains; and (g) the Genome Context tool that shows the gene neighborhood around the given COG for all genomes that encode proteins of the given COG.
The COG data set and the COGnitor program also are available by anonymous ftp at ftp://ftp.ncbi.nih.gov/pub/COG.
Future Directions
Substantial evolution of the COGs is expected in the near future in terms of both growth by adding more genomes and the addition of new functionalities and layers of presentation. Quantitatively, the main forthcoming addition is the COGs for eukaryotic genomes, which are expected to approximately double the size of the COG system. Many of the COGs include paralogous proteins, and this will be addressed by introducing hierarchical organization into the COG system, whereby related COGs will be unified at a higher level. In addition, partial integration of the COGs with the NCBI's Conserved Domains Database (CDD) is expected (Chapter 3), which will result in a more flexible and informative representation of the domain organization of proteins and of structural information that is available for COG members.
The COG Team
The COG system is developed and maintained by a team of programmers and expert biologists.
Project leader: Eugene V. Koonin.
The programming group: Roman L. Tatusov (group leader), Boris Kiryutin, Victor Smirnov, and Alexander Sverdlov (student)
The annotation group: Darren A. Natale (group leader), Natalie Fedorova, Anastasia Nikolskaya, Aviva Jacobs, Jodie Yin, B. Sridhar Rao, Dmitri M. Krylov, Sergei Mekhedov, John Jackson, Raja Mazumder, and Sona Vasudevan
References
- 1.
- Fitch WM . Distinguishing homologous from analogous proteins. Syst Zool. 1970;19:99–106. [PubMed: 5449325]
- 2.
- Fitch WM . Homology: a personal view on some of the problems. Trends Genet. 2000;16:227–231. [PubMed: 10782117]
- 3.
- Tatusov RL , Koonin EV , Lipman DJ . A genomic perspective on protein families. Science. 1997;278:631–637. [PubMed: 9381173]
- 4.
- Tatusov RL , Galperin MY , Natale DA , Koonin EV . The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–36. [PMC free article: PMC102395] [PubMed: 10592175]
- 5.
- Tatusov RL , Natale DA , Garkavtsev IV , Tatusova TA , Shankavaram UT , Rao BS , Kiryutin B , Galperin MY , Fedorova ND , Koonin EV . The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001;29:22–28. [PMC free article: PMC29819] [PubMed: 11125040]
- 6.
- Altschul SF , Madden TL , Schaffer AA , Zhang J , Zhang Z , Miller W , Lipman DJ . Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. [PMC free article: PMC146917] [PubMed: 9254694]
- 7.
- Wootton JC , Federhen S . Analysis of compositionally biased regions in sequence databases. Methods Enzymol. 1996;266:554–571. [PubMed: 8743706]
- 8.
- Lupas A , Van Dyke M , Stock J . Predicting coiled coils from protein sequences. Science. 1991;252:1162–1164. [PubMed: 2031185]
- 9.
- Natale DA , Shankavaram UT , Galperin MY , Wolf YI , Aravind L , Koonin EV . Genome annotation using clusters of orthologous groups of proteins (COGs)—towards understanding the first genome of a Crenarchaeon Genome Biol in press. [PMC free article: PMC15027] [PubMed: 11178258]
- 10.
- Riley M . Functions of the gene products of Escherichia coli. Microbiol Rev. 1993;57:862–952. [PMC free article: PMC372942] [PubMed: 7508076]
- 11.
- Thompson JD , Higgins DG , Gibson TJ . CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. [PMC free article: PMC308517] [PubMed: 7984417]
- The Clusters of Orthologous Groups (COGs) Database: Phylogenetic Classification ...The Clusters of Orthologous Groups (COGs) Database: Phylogenetic Classification of Proteins from Complete Genomes - The NCBI Handbook
Your browsing activity is empty.
Activity recording is turned off.
See more...